Color normalization¶
The first step in analyzing digital pathology images is often preprocessing the color image to correct staining or imaging variations. These examples illustrate how to use HistomicsTK to normalize color profiles and to generate augmented color images for machine learning.
[1]:
import girder_client
import numpy as np
from skimage.transform import resize
from matplotlib import pylab as plt
from matplotlib.colors import ListedColormap
from histomicstk.preprocessing.color_normalization import reinhard
from histomicstk.saliency.tissue_detection import (
get_slide_thumbnail, get_tissue_mask)
from histomicstk.annotations_and_masks.annotation_and_mask_utils import (
get_image_from_htk_response)
from histomicstk.preprocessing.color_normalization.\
deconvolution_based_normalization import deconvolution_based_normalization
from histomicstk.preprocessing.color_deconvolution.\
color_deconvolution import color_deconvolution_routine, stain_unmixing_routine
from histomicstk.preprocessing.augmentation.\
color_augmentation import rgb_perturb_stain_concentration, perturb_stain_concentration
Start girder client and set analysis parameters¶
[2]:
APIURL = 'http://candygram.neurology.emory.edu:8080/api/v1/'
SAMPLE_SLIDE_ID = '5d817f5abd4404c6b1f744bb'
gc = girder_client.GirderClient(apiUrl=APIURL)
# gc.authenticate(interactive=True)
gc.authenticate(apiKey='kri19nTIGOkWH01TbzRqfohaaDWb6kPecRqGmemb')
MAG = 1.0
# color norm. standard (from TCGA-A2-A3XS-DX1, Amgad et al, 2019)
cnorm = {
'mu': np.array([8.74108109, -0.12440419, 0.0444982]),
'sigma': np.array([0.6135447, 0.10989545, 0.0286032]),
}
# TCGA-A2-A3XS-DX1_xmin21421_ymin37486_.png, Amgad et al, 2019)
# for macenco (obtained using rgb_separate_stains_macenko_pca()
# and reordered such that columns are the order:
# Hamtoxylin, Eosin, Null
W_target = np.array([
[0.5807549, 0.08314027, 0.08213795],
[0.71681094, 0.90081588, 0.41999816],
[0.38588316, 0.42616716, -0.90380025],
])
# visualization color map
vals = np.random.rand(256, 3)
vals[0, ...] = [0.9, 0.9, 0.9]
cMap = ListedColormap(1 - vals)
# for visualization
ymin, ymax, xmin, xmax = 1000, 1500, 2500, 3000
# for reproducibility
np.random.seed(0)
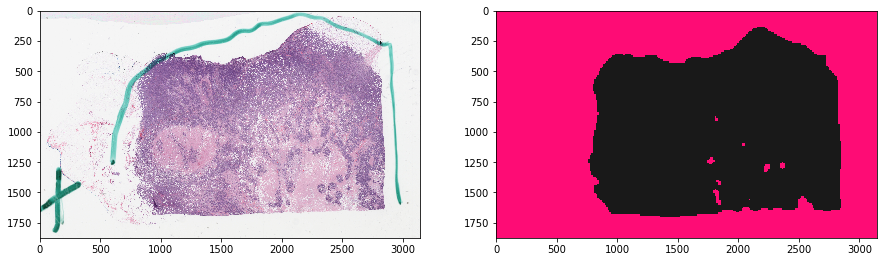
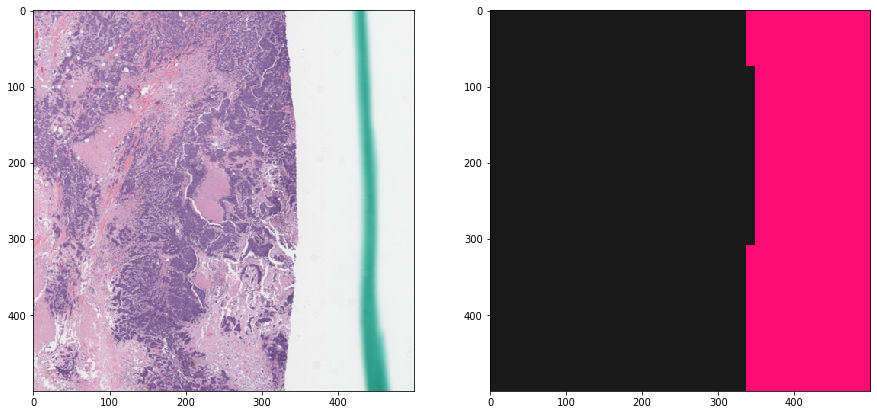
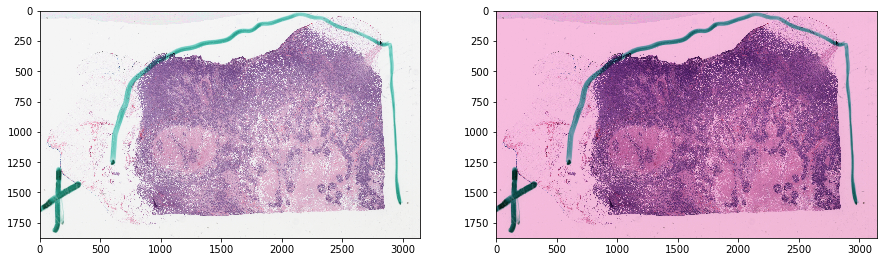
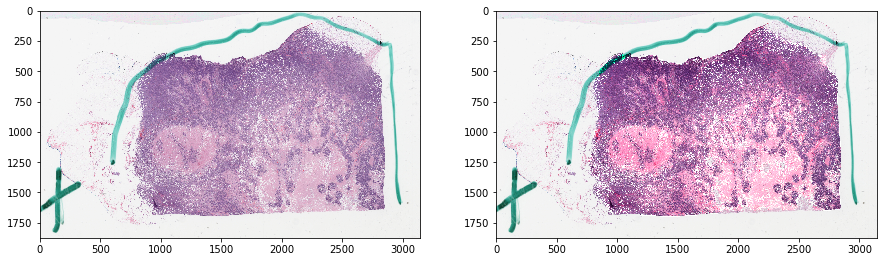
Get images and tissue mask¶
[3]:
# get RGB image at a small magnification
slide_info = gc.get('item/%s/tiles' % SAMPLE_SLIDE_ID)
getStr = '/item/%s/tiles/region?left=%d&right=%d&top=%d&bottom=%d' % (
SAMPLE_SLIDE_ID, 0, slide_info['sizeX'], 0, slide_info['sizeY'],
) + '&magnification=%.2f' % MAG
tissue_rgb = get_image_from_htk_response(
gc.get(getStr, jsonResp=False))
# get mask of things to ignore
thumbnail_rgb = get_slide_thumbnail(gc, SAMPLE_SLIDE_ID)
mask_out, _ = get_tissue_mask(
thumbnail_rgb, deconvolve_first=True,
n_thresholding_steps=1, sigma=1.5, min_size=30)
mask_out = resize(
mask_out == 0, output_shape=tissue_rgb.shape[:2],
order=0, preserve_range=True) == 1
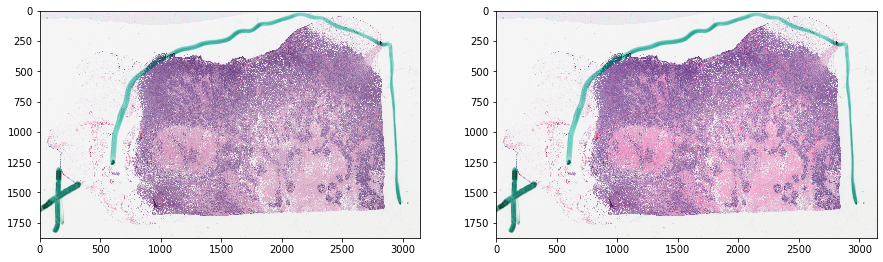
Let’s visualize the data¶
[4]:
f, ax = plt.subplots(1, 2, figsize=(15, 15))
ax[0].imshow(tissue_rgb)
ax[1].imshow(mask_out, cmap=cMap)
plt.show()
f, ax = plt.subplots(1, 2, figsize=(15, 15))
ax[0].imshow(tissue_rgb[ymin:ymax, xmin:xmax, :])
ax[1].imshow(mask_out[ymin:ymax, xmin:xmax], cmap=cMap)
plt.show()
Reinhard normalization¶
[5]:
print(reinhard.__doc__)
Perform Reinhard color normalization.
Transform the color characteristics of an image to a desired standard.
The standard is defined by the mean and standard deviations of the target
image in LAB color space defined by Ruderman. The input image is converted
to Ruderman's LAB space, the LAB channels are each centered and scaled to
zero-mean unit variance, and then rescaled and shifted to match the target
image statistics. If the LAB statistics for the input image are provided
(`src_mu` and `src_sigma`) then these will be used for normalization,
otherwise they will be derived from the input image `im_src`.
Parameters
----------
im_src : array_like
An RGB image
target_mu : array_like
A 3-element array containing the means of the target image channels
in LAB color space.
target_sigma : array_like
A 3-element array containing the standard deviations of the target
image channels in LAB color space.
src_mu : array_like, optional
A 3-element array containing the means of the source image channels in
LAB color space. Used with reinhard_stats for uniform normalization of
tiles from a slide.
src_sigma : array, optional
A 3-element array containing the standard deviations of the source
image channels in LAB color space. Used with reinhard_stats for
uniform normalization of tiles tiles from a slide.
mask_out : array_like, default is None
if not None, should be (m, n) boolean numpy array.
This method uses numpy masked array functionality to only use
non-masked areas in calculations. This is relevant because elements
like blood, sharpie marker, white space, etc would throw off the
reinhard normalization by affecting the mean and stdev. Ideally, you
want to exclude these elements from both the target image (from which
you calculate target_mu and target_sigma) and from the source image
to be normalized.
Returns
-------
im_normalized : array_like
Color Normalized RGB image
See Also
--------
histomicstk.preprocessing.color_conversion.rgb_to_lab,
histomicstk.preprocessing.color_conversion.lab_to_rgb
References
----------
.. [#] E. Reinhard, M. Adhikhmin, B. Gooch, P. Shirley, "Color transfer
between images," in IEEE Computer Graphics and Applications, vol.21,
no.5,pp.34-41, 2001.
.. [#] D. Ruderman, T. Cronin, and C. Chiao, "Statistics of cone responses
to natural images: implications for visual coding," J. Opt. Soc. Am. A
vol.15, pp.2036-2045, 1998.
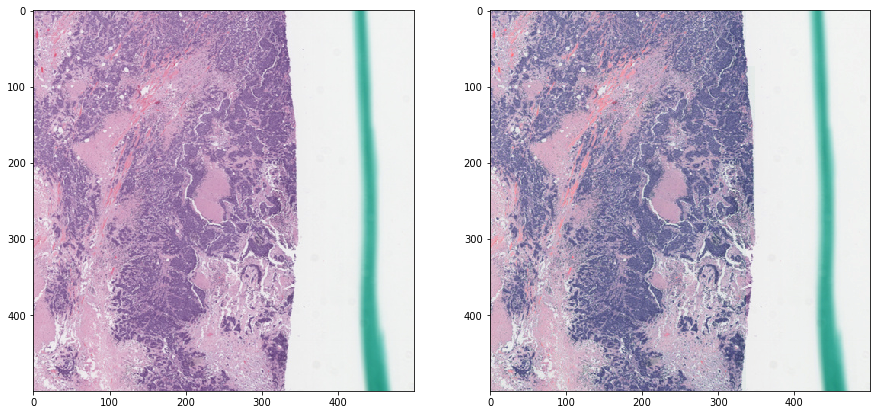
Reinhard normalization - without masking¶
Notice how non-tissue elements throw off the normalization algorithm.
[6]:
tissue_rgb_normalized = reinhard(
tissue_rgb, target_mu=cnorm['mu'], target_sigma=cnorm['sigma'])
[7]:
def vis_result():
f, ax = plt.subplots(1, 2, figsize=(15, 15))
ax[0].imshow(tissue_rgb)
ax[1].imshow(tissue_rgb_normalized)
plt.show()
f, ax = plt.subplots(1, 2, figsize=(15, 15))
ax[0].imshow(tissue_rgb[ymin:ymax, xmin:xmax, :])
ax[1].imshow(tissue_rgb_normalized[ymin:ymax, xmin:xmax, :])
plt.show()
vis_result()
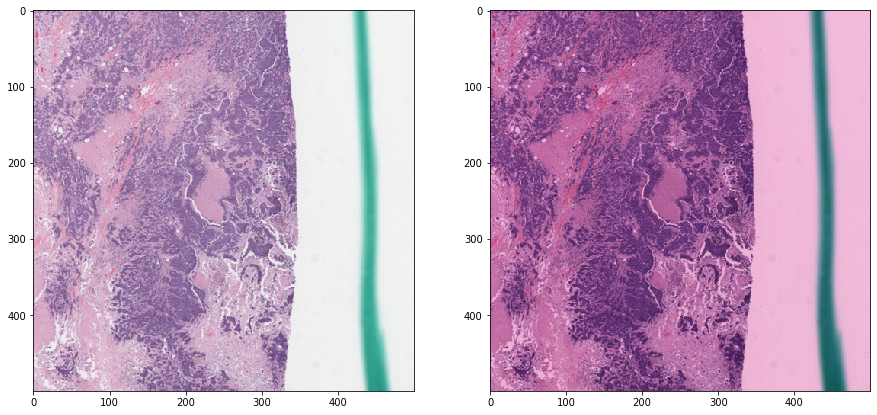
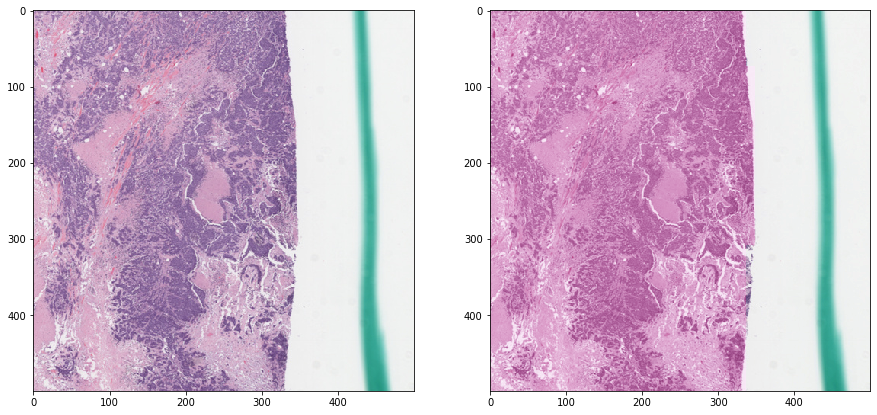
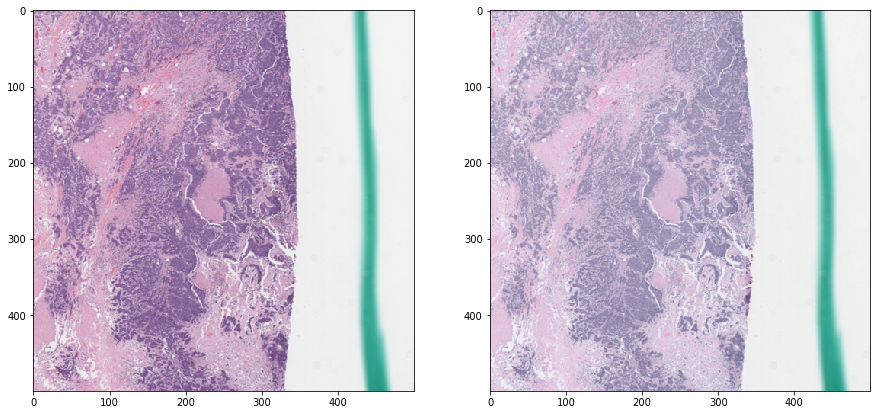
Reinhard normalization - with masking¶
Now we mask out irrelevant areas when calculating the statistics. Notice how the result is much better.
[8]:
tissue_rgb_normalized = reinhard(
tissue_rgb, target_mu=cnorm['mu'], target_sigma=cnorm['sigma'],
mask_out=mask_out)
[9]:
vis_result()
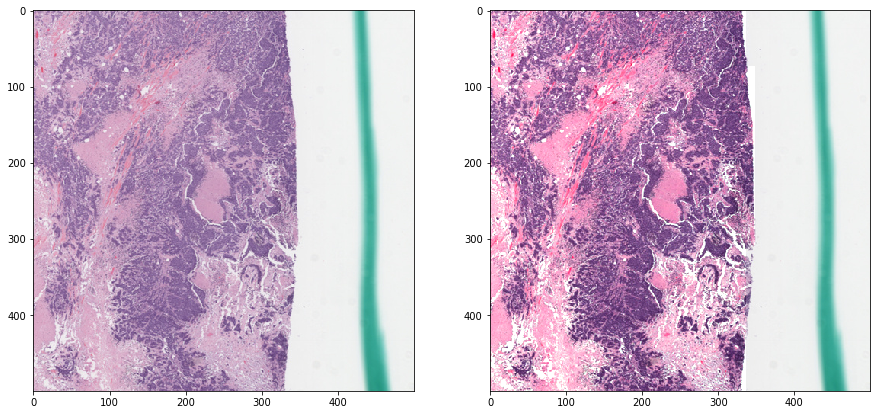
Deconvolution-based normalization¶
Unlike reinhard, which simply matched the mean and standard deviation of the image to a prespecified target, these methods are “smarter”, in the sense that they first unmix the stains and then convolve with a desired stain standard.
Macenko stain unmixing is used by default, but the method is general and may be used with other stain unmixing methods in this repository such as the SNMF method of Xu et al.
[10]:
print(deconvolution_based_normalization.__doc__)
Perform color normalization using color deconvolution to transform the.
... color characteristics of an image to a desired standard.
After the image is deconvolved into its component stains (eg, H&E), it is
convolved with a stain column vectors matrix from the target image from
which the color characteristics need to be transferred.
Parameters
------------
im_src : array_like
An RGB image (m x n x 3) to color normalize
W_source : np array, default is None
A 3x3 matrix of source stain column vectors. Only provide this
if you know the stains matrix in advance (unlikely) and would
like to perform supervised deconvolution. If this is not provided,
stain_unmixing_routine() is used to estimate W_source.
W_target : np array, default is None
A 3x3 matrix of target stain column vectors. If not provided,
and im_target is also not provided, the default behavior is to use
histomicstk.preprocessing.color_deconvolution.stain_color_map
to provide an idealized target matrix.
im_target : array_like, default is None
An RGB image (m x n x 3) that has good color properties that ought to
be transferred to im_src. If you provide this parameter, im_target
will be used to extract W_target and the W_target parameter will
be ignored.
stains : list, optional
List of stain names (order is important). Default is H&E. This is
particularly relevant in macenco where the order of stains is not
preserved during stain unmixing, so this method uses
histomicstk.preprocessing.color_deconvolution.find_stain_index
to reorder the stains matrix to the order provided by this parameter
mask_out : array_like, default is None
if not None, should be (m x n) boolean numpy array.
This parameter ensures exclusion of non-masked areas from calculations
and normalization. This is relevant because elements like blood,
sharpie marker, white space, etc may throw off the normalization.
stain_unmixing_routine_params : dict, default is empty dict
k,v for stain_unmixing_routine().
Returns
--------
array_like
Color Normalized RGB image (m x n x 3)
See Also
--------
histomicstk.preprocessing.color_deconvolution.color_deconvolution_routine
histomicstk.preprocessing.color_convolution.color_convolution
References
----------
.. [#] Van Eycke, Y. R., Allard, J., Salmon, I., Debeir, O., &
Decaestecker, C. (2017). Image processing in digital pathology: an
opportunity to solve inter-batch variability of immunohistochemical
staining. Scientific Reports, 7.
.. [#] Macenko, M., Niethammer, M., Marron, J. S., Borland, D.,
Woosley, J. T., Guan, X., ... & Thomas, N. E. (2009, June).
A method for normalizing histology slides for quantitative analysis.
In Biomedical Imaging: From Nano to Macro, 2009. ISBI'09.
IEEE International Symposium on (pp. 1107-1110). IEEE.
.. [#] Xu, J., Xiang, L., Wang, G., Ganesan, S., Feldman, M., Shih, N. N.,
...& Madabhushi, A. (2015). Sparse Non-negative Matrix Factorization
(SNMF) based color unmixing for breast histopathological image
analysis. Computerized Medical Imaging and Graphics, 46, 20-29.
[11]:
print(color_deconvolution_routine.__doc__)
Unmix stains mixing followed by deconvolution (wrapper).
Parameters
------------
im_rgb : array_like
An RGB image (m x n x 3) to colro normalize
W_source : np array, default is None
A 3x3 matrix of source stain column vectors. Only provide this
if you know the stains matrix in advance (unlikely) and would
like to perform supervised deconvolution. If this is not provided,
stain_unmixing_routine() is used to estimate W_source.
kwargs : k,v pairs
Passed as-is to stain_unmixing_routine() if W_source is None.
Returns
--------
Output from color_deconvolution()
See Also
--------
histomicstk.preprocessing.color_deconvolution.stain_unmixing_routine
histomicstk.preprocessing.color_deconvolution.color_deconvolution
[12]:
print(stain_unmixing_routine.__doc__)
Perform stain unmixing using the method of choice (wrapper).
Parameters
------------
im_rgb : array_like
An RGB image (m x n x 3) to unmix.
stains : list, optional
List of stain names (order is important). Default is H&E. This is
particularly relevant in macenco where the order of stains is not
preserved during stain unmixing, so this method uses
histomicstk.preprocessing.color_deconvolution.find_stain_index
to reorder the stains matrix to the order provided by this parameter
stain_unmixing_method : str, default is 'macenko_pca'
stain unmixing method to use. It should be one of the following
'macenko_pca', or 'xu_snmf'.
stain_unmixing_params : dict, default is an empty dict
kwargs to pass as-is to the stain unmixing method.
mask_out : array_like, default is None
if not None, should be (m x n) boolean numpy array.
This parameter ensures exclusion of non-masked areas from calculations
and normalization. This is relevant because elements like blood,
sharpie marker, white space, etc may throw off the normalization.
Returns
--------
Wc : array_like
A 3x3 complemented stain matrix.
See Also
--------
histomicstk.preprocessing.color_deconvolution.separate_stains_macenko_pca
histomicstk.preprocessing.color_deconvolution.separate_stains_xu_snmf
References
----------
.. [#] Macenko, M., Niethammer, M., Marron, J. S., Borland, D.,
Woosley, J. T., Guan, X., ... & Thomas, N. E. (2009, June).
A method for normalizing histology slides for quantitative analysis.
In Biomedical Imaging: From Nano to Macro, 2009. ISBI'09.
IEEE International Symposium on (pp. 1107-1110). IEEE.
.. [#] Xu, J., Xiang, L., Wang, G., Ganesan, S., Feldman, M., Shih, N. N.,
...& Madabhushi, A. (2015). Sparse Non-negative Matrix Factorization
(SNMF) based color unmixing for breast histopathological image
analysis. Computerized Medical Imaging and Graphics, 46, 20-29.
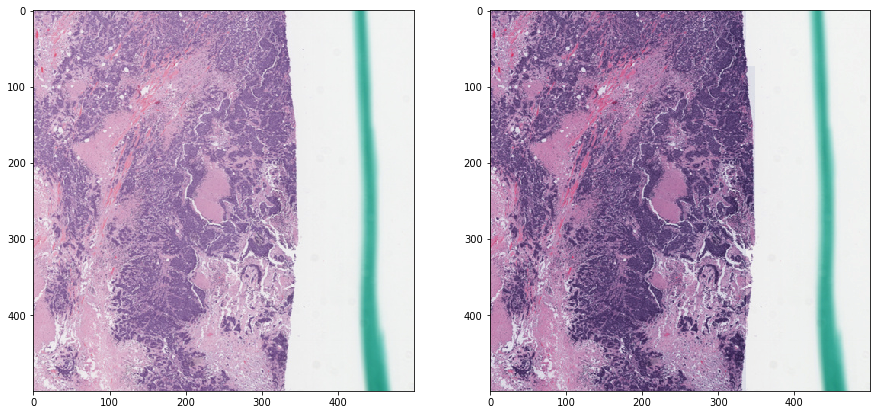
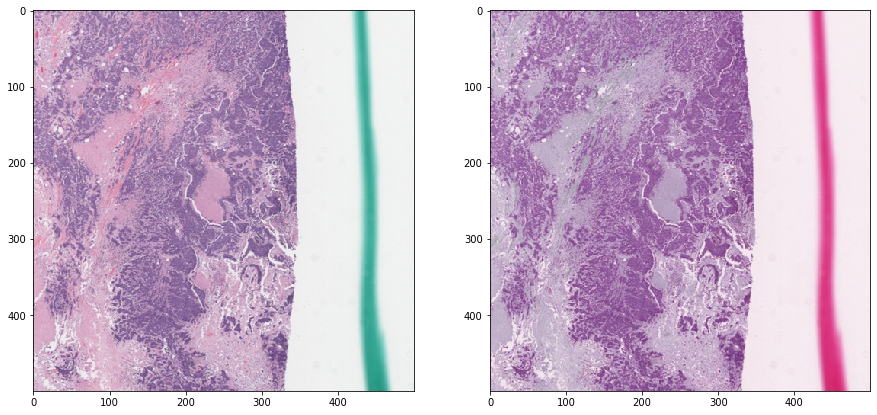
Macenko normalization - without masking¶
[13]:
stain_unmixing_routine_params = {
'stains': ['hematoxylin', 'eosin'],
'stain_unmixing_method': 'macenko_pca',
}
tissue_rgb_normalized = deconvolution_based_normalization(
tissue_rgb, W_target=W_target,
stain_unmixing_routine_params=stain_unmixing_routine_params)
[14]:
vis_result()
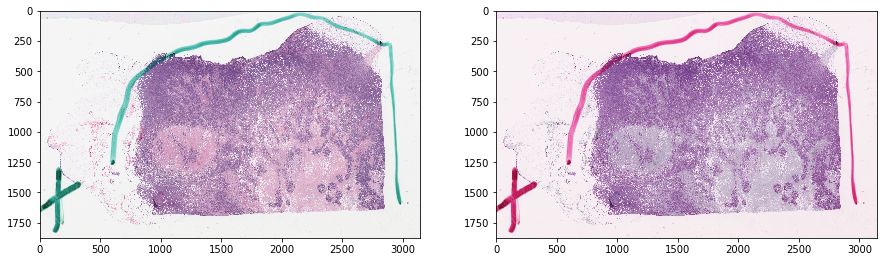
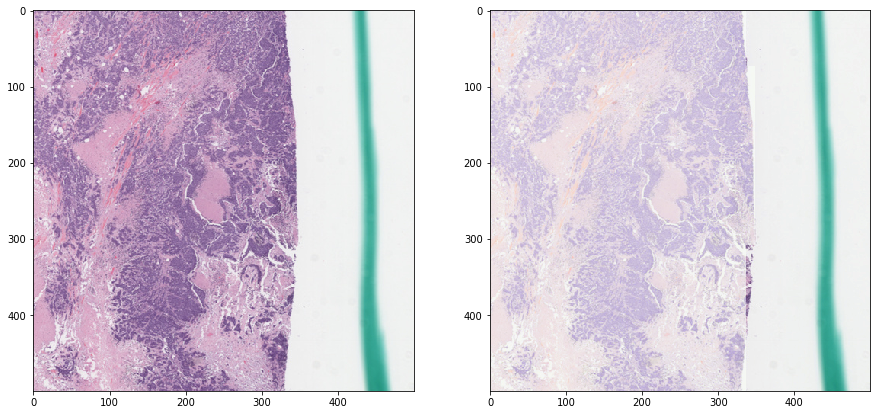
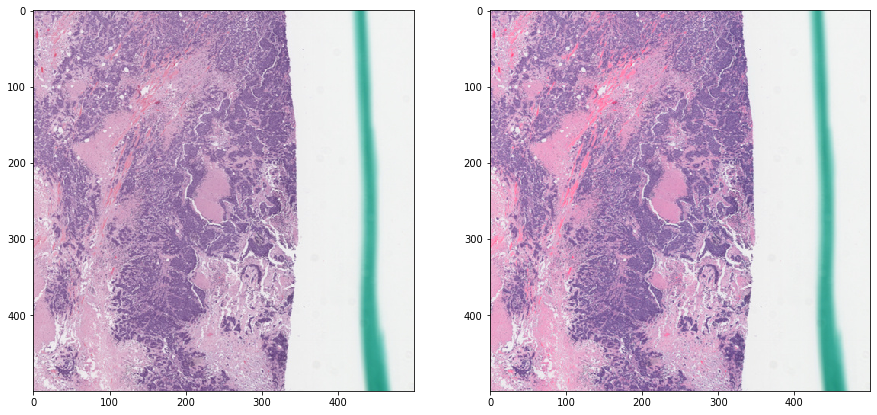
Macenko normalization - with masking¶
[15]:
tissue_rgb_normalized = deconvolution_based_normalization(
tissue_rgb, W_target=W_target,
stain_unmixing_routine_params=stain_unmixing_routine_params,
mask_out=mask_out)
[16]:
vis_result()
“Smart” color augmentation¶
This is an implementation of the paper by Tellez et al, 2018 (see docstring below), whereby the stain concentrations are perturbed so that a more realistic model of stiaining variability in histology is modeled.
[17]:
print(perturb_stain_concentration.__doc__)
Perturb stain concentrations in SDA space and return augmented image.
This is an implementation of the method described in Tellez et
al, 2018 (see below). The SDA matrix is perturbed by multiplying each
channel independently by a value chosen from a random uniform distribution
in the range [1 - sigma1, 1 + sigma1], then add a value chose from another
random uniform distribution in the range [-sigma2, sigma2].
Parameters
------------
StainsFloat : array_like
An intensity image (m, n, 3) of deconvolved stains that is unbounded,
suitable for reconstructing color images of deconvolved stains
with color_convolution.
W : array_like
A 3x3 complemented stain matrix.
I_0 : float or array_like, optional
A float a 3-vector containing background RGB intensities.
If unspecified, use the old OD conversion.
mask_out : array_like, default is None
if not None, should be (m x n) boolean numpy array.
This parameter ensures exclusion of non-masked areas from perturbing.
This is relevant because elements like blood, sharpie marker,
white space, etc cannot be simply modeled as a mix of two stains.
sigma1 : float
parameter, see beginning of this docstring.
sigma2 : float
parameter, see beginning of this docstring.
Returns
--------
array_like
Color augmented RGB image (m x n x 3)
References
----------
.. [#] Tellez, David, Maschenka Balkenhol, Irene Otte-Höller,
Rob van de Loo, Rob Vogels, Peter Bult, Carla Wauters et al.
"Whole-slide mitosis detection in H&E breast histology using PHH3
as a reference to train distilled stain-invariant convolutional
networks." IEEE transactions on medical imaging 37, no. 9
(2018): 2126-2136.
.. [#] Tellez, David, Geert Litjens, Peter Bandi, Wouter Bulten,
John-Melle Bokhorst, Francesco Ciompi, and Jeroen van der Laak.
"Quantifying the effects of data augmentation and stain color
normalization in convolutional neural networks for computational
pathology." arXiv preprint arXiv:1902.06543 (2019).
.. [#] Implementation inspired by Peter Byfield StainTools repository. See
https://github.com/Peter554/StainTools/blob/master/LICENSE.txt
for copyright license (MIT license).
[18]:
print(rgb_perturb_stain_concentration.__doc__)
Apply wrapper that calls perturb_stain_concentration() on RGB.
Parameters
------------
im_rgb : array_like
An RGB image (m x n x 3) to colro normalize
stain_unmixing_routine_params : dict
kwargs to pass as-is to the color_deconvolution_routine().
kwargs : k,v pairs
Passed as-is to perturb_stain_concentration()
Returns
--------
array_like
Color augmented RGB image (m x n x 3)
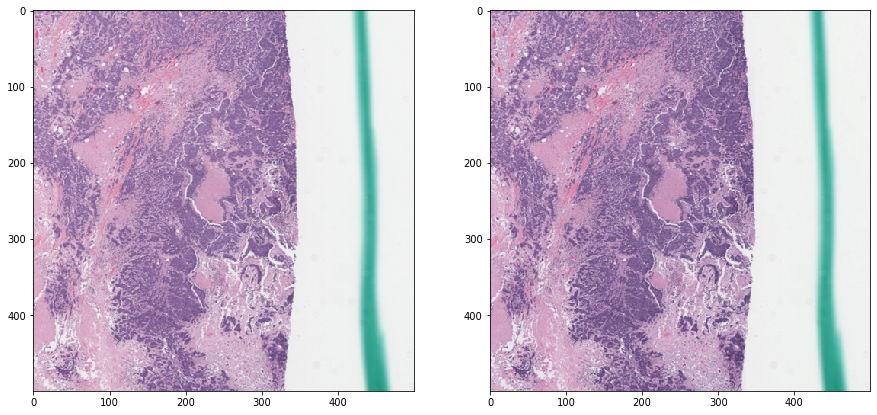
Let’s perturb the H&E concentrations a bit¶
[19]:
rgb = tissue_rgb[ymin:ymax, xmin:xmax, :]
exclude = mask_out[ymin:ymax, xmin:xmax]
augmented_rgb = rgb_perturb_stain_concentration(rgb, mask_out=exclude)
[20]:
def vis_augmentation():
f, ax = plt.subplots(1, 2, figsize=(15, 15))
ax[0].imshow(rgb)
ax[1].imshow(augmented_rgb)
plt.show()
vis_augmentation()
Try a few more times¶
[21]:
for _ in range(5):
augmented_rgb = rgb_perturb_stain_concentration(rgb, mask_out=exclude)
vis_augmentation()